技術紹介
量子化学分子モデリングとは?
- 表面電子密度の汎関数理論を用いて分子ファンデルワールス力集合系の化学現象の検証・予測すること
- 分子ファンデルワールス力集合系のエネルギー構造は,電磁波エネルギー吸収スペクトル(UV-Vis, NIR, IR, FIR, NMR, Raman spectra)で可視化できること
主な手順は次の通り。
- 水中,水素(H2)とヒドロキシルラジカル(HO.) の分子会合で何が起こるかを,量子化学分子モデリングでの解明を試みた。 会合体[H2&HO. &H2O]の生成は,水素結合やイオン双極子相互作用を総称するvan der Waals &Coulomb interactions (以後vdWと略する)力に基づく。そのような分子会合体が水系を構成すると考える。
- 3量体分子分子集団を量子化学分子モデリングすなわち,密度汎関数理論に基づく分子モデリング(DFT/MM)は、かってのスパーコンピュータレベルの能力に達したパーソナルコンピューター(PC)を用いて行うことで可能である。すなわち、Wavefunction Inc/Q-chem Incが開発したSpartan 18を用いることで,誰でもがvdW会合分子間の結合に関与する電子軌道の数理的解析を行うことができる。
- 会合体の立体構造を求めることは、反応の遷移状態や反応中間体を検証・予見することにつながる。分子会合体の分子力学法(MMFF)で求められるエネルギー構造をsingle point計算すると,反応遷移構造を知ることになる(事例1−1) 。また,DFT/MMでえられる平衡立体構造(equilibrium geometry, EQG)は,安定な反応中間体構造に相当する( 事例1ー2) 。
- DFT/MMでは,分子会合体の分子軌道の持つエネルギー構造を電磁波エネルギー吸収スペクトルクで可視化する。そのスペクトルは,理論スペクトルと呼ぶことができる。実測電磁波スペクトルクと理論電磁波スペクトルを比較することで,会合体構造の検証・予見が可能となる(事例2) 。
- 電磁波エネルギー吸収スペクトルクとは、UV/Vis, Near IR, IR, FIR,MW, Radio wave, NMR 吸収スペクトルを指す。DFT/MMで求められる理論UV/Visでは,吸収極大に対して最高占有軌道群(HOMO)と 最低空軌道群(LUMO)間の遷移の帰属とその強度を示す遷移確率を知ることができる。
- ラジカルや不対電子をもつ無機・有機分子の会合体もDFT/MMでの解析が可能である(事例3) 。
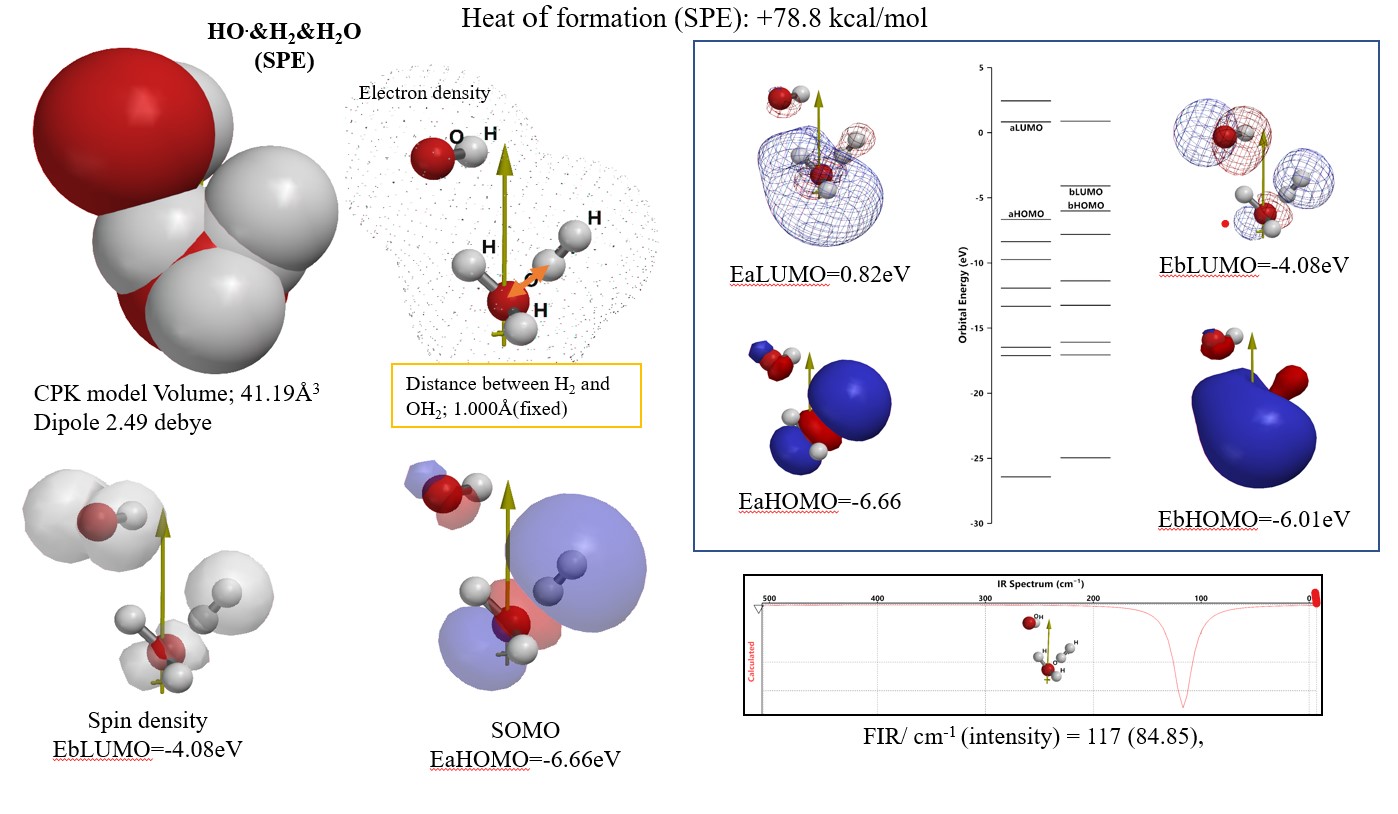
量子化学分子モデリングの事例1-1:Single point energy calculationで求めた会合体構造 HO.&H2&H2O(SPE)は,反応遷移状態とみなすことができる。
水素分子がヒドロキシルラジカルと体内細胞環境で反応するかの疑問を解くために,HO radical とH2なびに H2Oのvan der Waals 会合体を分子力学手法でその構造を決定する(下図)。その構造に対するsingle point energyを求めた。その時に求められたHeat of formation (SPE)は,吸熱的で +77.8 kcal/molと求められる。分子軌道のエネルギー構造(EbLUMO, EbHOMO)は,その構造が還元されやすく,酸化もされやすいことを検証する。理論FIR spectrumからTHz領域の電磁波でその分子会合反応が,発熱的に進行することが検証できた。
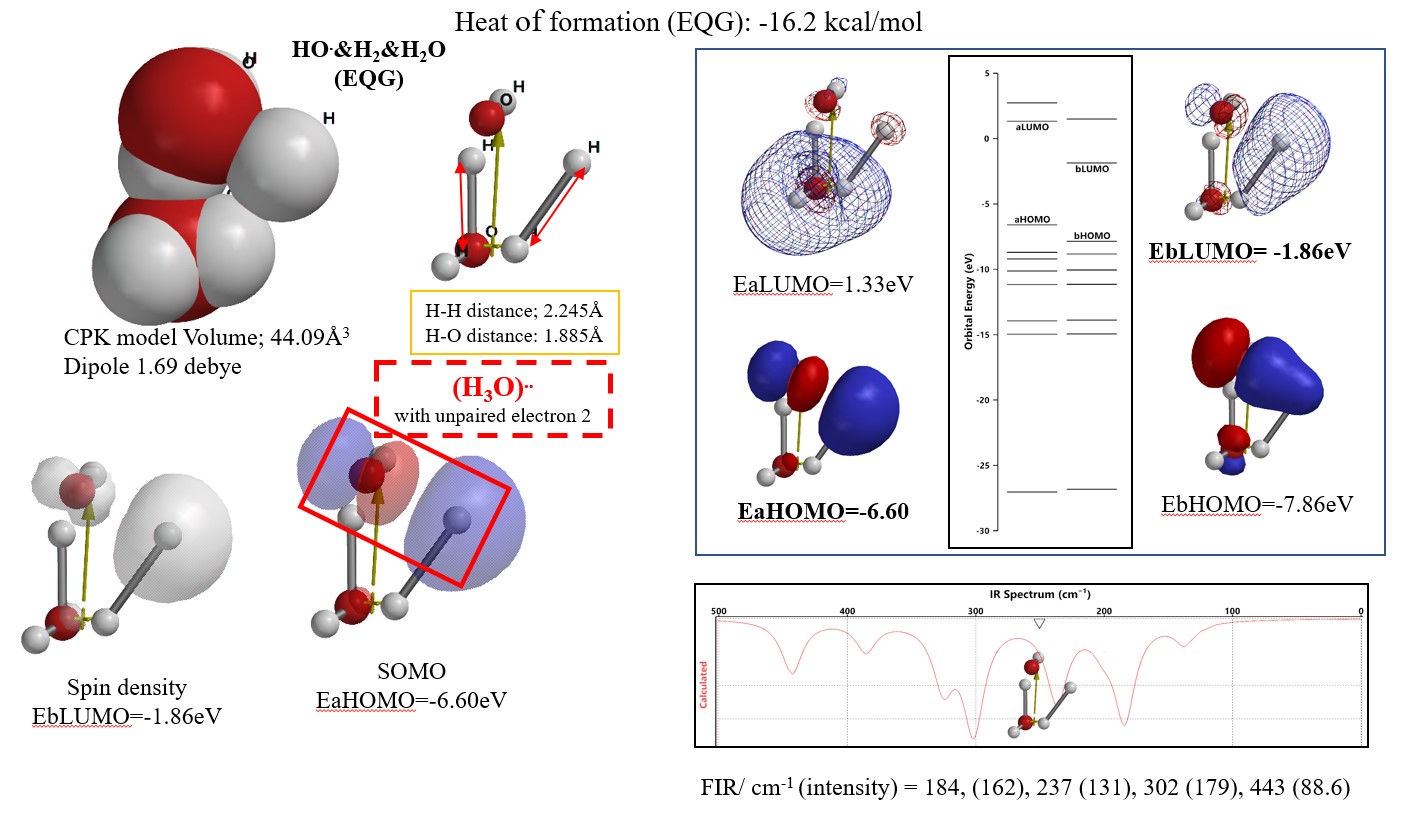
量子化学分子モデリングの事例1-2:
Equilibrium geometry 会合体構造[HO.&H2&H2O (EQG)]は,開裂状態水素分子(H2O)がHO.に対して還元力を示す反応中間体とみなすことができる。
HO radical と H2と H2Oのvan der Waals 会合体のSPE構造に対するEquilibrium geometry (EQG)構造を求めた。その時に求められたHeat of formation (EQG)は,発熱的で-16.2 kcal/molである。EQG構造は,水素分子が開裂状態にあり,理論FIR spectrumから電磁波エネルギーの吸収によって反応が発熱的に進行することが示された。開裂状態の水素分子とヒドロキシルラジカルとの会合中間体 [(H3O)..]の生成が予見できる。
量子化学分子モデリングの事例2:
ラジカル分子の理論UV-Vis spectrumが測定UV-Vis spectrumと一致

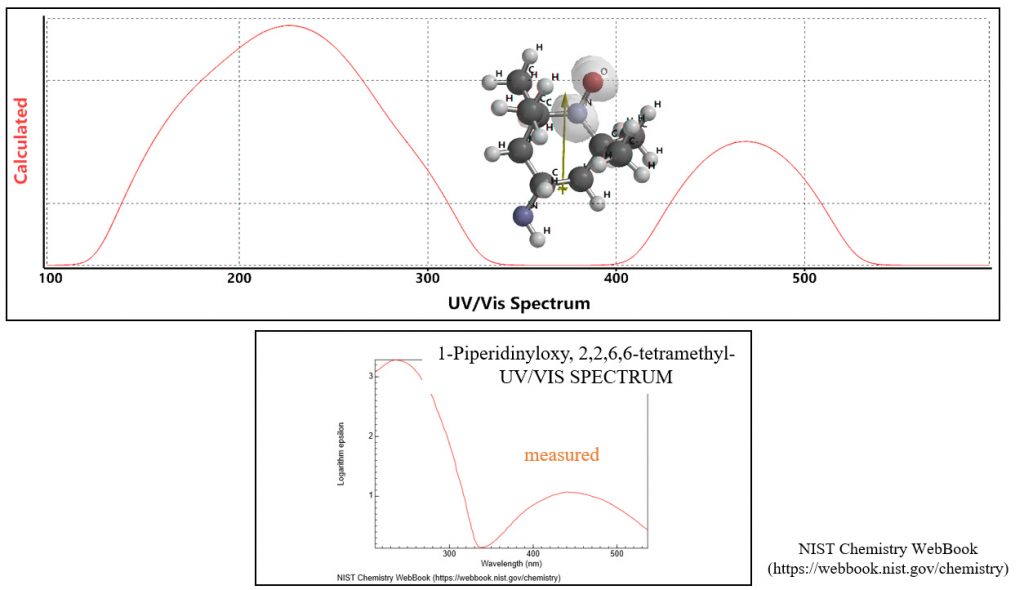
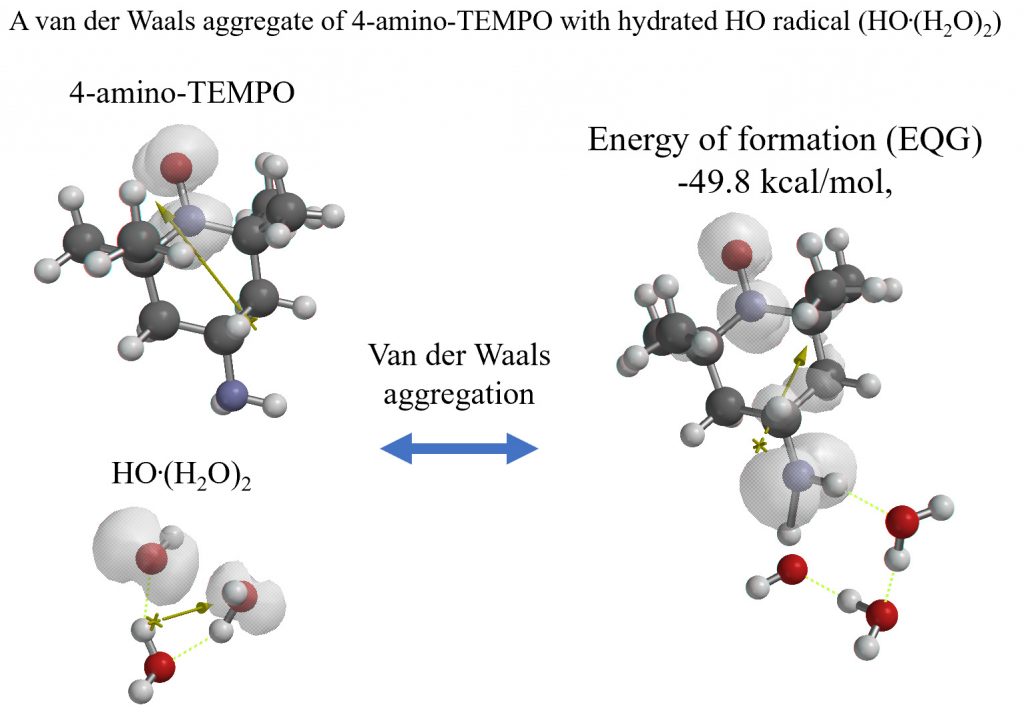
量子化学分子モデリングの事例3:
ヒドロキシルラジカルはラジカルトラップ剤4-amino-TEMPOと反応してラジカルサイトを安定化することの検証